
〜 京都大学放射線生物研究センターが発行する「放生研ニュース」に寄稿したミニレビューを転載します〜
はじめに
呼吸をする、あるいは、食べ物を飲み込む、といった生命活動に欠かすことができない身体の動きは、脳が「運動ニューロン」と呼ばれる神経細胞を介して、骨格筋の伸び縮みをコントロールすることで生み出されている。筋萎縮性側索硬化症(Amyotrophic lateral sclerosis, ALS)は、運動ニューロンが徐々に変性して機能を失い、やがて筋萎縮による致命的な麻痺に至る神経変性疾患である。本邦では、現在およそ9,000人がこの難病を患っていると推計されている。ALS全体の5~10%は単一の遺伝子座の変異に連鎖する家族性ALSであり、1993年に、銅・亜鉛-スーパーオキシドディスムターゼをコードするSOD1遺伝子が最初に同定されて以来、20種類以上のALSの原因遺伝子が同定されている。家族性ALSの発症メカニズムは、その原因遺伝子の生物学的機能を出発点とする研究が主流であるが、運動ニューロンの変性過程を十分説明するには至っていない。一方、ALSの大部分(90~95%)は、単一の原因変異が同定されない孤発性である。孤発性ALSは、その原因を特定の遺伝子機能の異常に遡ることが困難であり、発症メカニズムの研究は、家族性ALSよりもさらに困難である。このミニレビューでは、この困難さをなんとか克服し、孤発性ALSの発症メカニズムに迫ることを目指した筆者らの取り組みを、光遺伝学に基づいたゼブラフィッシュALSモデル[1]を中心に紹介したい。
光遺伝学を用いたALSモデル
孤発性ALSの運動ニューロンには、細胞質にユビキチン陽性の封入体が蓄積することが知られていたが、この封入体の主成分が、RNA/DNA結合タンパク質TDP-43の凝集体であることが2006年に見出された[2, 3]。この発見がブレイクスルーとなり、凝集したTDP-43が発揮する細胞毒性によって運動ニューロンが変性する、という病態仮説が提唱され、盛んに検証されてきた。一方で、この仮説は魅力的ではあるものの、細胞や動物モデルにおいて、ALS病態を反映したTDP-43の凝集体形成を再現することは容易ではなかった。例えば、TDP-43を過剰発現すると細胞質にTDP-43の凝集体が形成されることがあるが、TDP-43の過剰発現が細胞毒性を発揮しうることが知られているために、着目する表現型が過剰発現と凝集のいずれに起因するのか、を判定することが困難であった。結果として、TDP-43凝集体が毒性を発揮するのか、否か、という論争には決着がついてない。

ALSの最大の特徴は、運動ニューロンが選択的に失われることである。しかし、運動ニューロンが身体の深部を走行する複雑な形態をもった大きい細胞であるために、人体や哺乳類動物モデルでは、運動ニューロンの全体像を捉えることや、運動ニューロンにおけるALS関連因子のダイナミックな分子の振る舞いを研究することが難しい。筆者らは、この困難さを軽減する可能性をもった小型熱帯魚ゼブラフィッシュを用いて、ALSモデルの開発に取り組んできた。ゼブラフィッシュの稚魚は、身体組織の透明性が高いので、蛍光タンパク質を発現させることで、運動ニューロンの全体像や細胞内部の構造を、個体を生かしたまま観察することができる(図1)。ゼブラフィッシュには、30以上ある脊髄節のそれぞれに、半脊髄あたり60個ほどの運動ニューロンが存在する。まず筆者らは、独自に開発したゼブラフィッシュGal4/UAS遺伝子発現法[4]を利用して、この60個のうちの1つであるCaPと呼ばれる運動ニューロンだけにTDP-43を過剰発現することで、TDP-43を凝集させようと試みた。ところが、TDP-43の過剰発現によって運動ニューロンの軸索の伸長は著しく阻害されたが、過剰発現されたTDP-43は細胞核に局在し、細胞質で凝集体は形成されなかった。この結果は、TDP-43の過剰発現が、細胞質で凝集することなく細胞毒性を発揮していることを示していた。したがって筆者らは、過剰発現によってTDP-43凝集を誘導して毒性を評価する、というアプローチを断念せざるを得なかった。
次に筆者らは、TDP-43の細胞内濃度の変動に依存することなく、TDP-43の凝集を誘導する手法の開発を模索した。筆者らが着目したのは、青い光を吸収すると凝集するシロイヌナズナのCryptochrome (CRY2)である。光凝集性を高める変異が導入されたCRY2(Cry2olig)[5]を、天然変性領域(Intrinsically Disordered Region, IDR)が存在するTDP-43のC末端側に融合させた光遺伝学型TDP-43(opTDP-43)を構築した(図2)[1]。TDP-43の凝集には、IDRを介した相転移が重要な役割を担っていると予想し、Cry2oligモジュールが青色光を吸収して集合すると、IDR間の物理的相互作用が促進され、opTDP-43が相転移を起こすのではないか、と考えた。
opTDP-43が、光照変換型TDP-43として機能するならば、まず暗所においては、TDP-43としての機能を保持していなくてはならない。このことを検証するために、TDP-43遺伝子を破壊した1細胞期のゼブラフィッシュ胚に、opTDP-43のメッセンジャーRNAを微量注入し、暗所で発生させた。その結果、TDP-43遺伝子破壊がもたらす血液循環の欠損がレスキューされたことから、opTDP-43は、暗所ではTDP-43として機能することがわかった。次に、青色光の照射によってopTDP-43が凝集体を形成するか、否か、について検証した。細胞のサイズが大きいために細胞核と細胞質を明確に区別できる骨格筋細胞にopTDP-43を発現させ、青色光を照射した。すると、細胞核に局在していたopTDP-43は、青色光の照射に伴って、徐々に細胞質にユビキチン陽性の凝集体を形成することがわかった。これらの結果から、opTDP-43は、暗所ではTDP-43として機能し、青色光を吸収すると凝集する、光変換型TDP-43であると結論づけた。当初、筆者らは、生体内観察が容易であるという理由で、上皮細胞や発生後期の筋細胞を用いて実験を行なっていたが、光照射に依存したopTDP-43の凝集を全く検出できなかった。これらの結果に落胆し、このプロジェクトを破棄してしまうところであったが、発生初期の筋細胞ではopTDP-43の凝集が起こることに、運良く気づくことができた。このことは、TDP-43の相転移や細胞内局在の制御が、細胞種や発生ステージによって異なることを示しており、生体内の運動ニューロンを用いた研究によってのみ解明できるTDP-43の特性があることを暗示している。
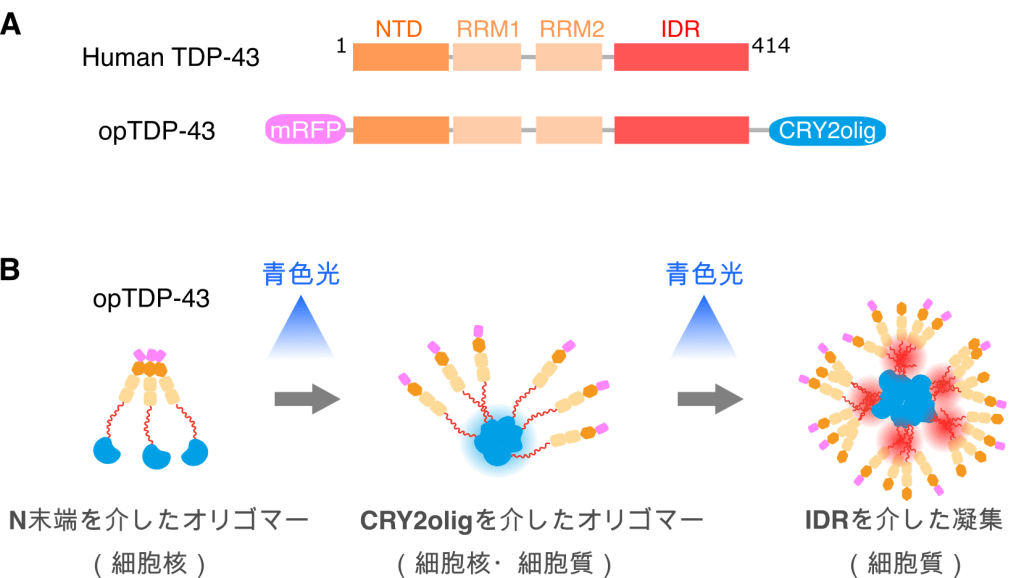
では、運動ニューロンでは、opTDP-43はどのように振る舞うのであろうか?筆者らは、運動ニューロンの発生を制御するホメオボックス遺伝子mnr2bのシス配列を利用し、Gal4/UAS法を用いてほぼ全ての運動ニューロンにopTDP-43を発現するフィッシュ系統を作成した。この魚に3時間にわたって青色光を照射すると、細胞核に局在していたopTDP-43が、徐々に細胞核と細胞質を含む細胞全体に拡散することがわかった。さらに、青色光照射を停止し、暗所に戻して飼育を続けると、意外なことに、opTDP-43の凝集体は形成されず、opTDP-43の細胞核への局在が回復していた。この一過的なopTDP-43の細胞全体への拡散が運動ニューロンに及ぼす効果を評価する為に、青色光を照射した運動ニューロンと、暗所で生育した運動ニューロンの細胞形態を詳しく比較した。その結果、3時間の青色光照射を施した運動ニューロンでは、神経軸索の総長が低下していた。この結果は、細胞質に凝集体として蓄積する前に、opTDP-43が既に細胞毒性を発揮していることを示している。本稿では割愛するが、その後筆者らは、遊泳魚に青色LEDを光照射するアリーナをセットアップし、数日間にわたって青色光を照射し続けると、ALS病態を模倣するopTDP-43凝集体が細胞質に形成されることを突き止めた。
本研究はTDP-43の凝集体が発揮する細胞毒性を検証することを当初の目的としたが、opTDP-43を光操作によって相転移させると、明確な凝集体が蓄積する前に、既に運動ニューロンが障害を受けている、という予想外の結果を得た。この結果は、残念ながらこの光遺伝学ALSモデルでは、TDP-43凝集の毒性を評価する、という当初の目的が達成できなことを示していた。一方、運動ニューロンにおけるTDP-43凝集の上流のイベントの解明は、ALSの原因究明にむけた非常に重要な研究課題であるが、現在、この課題にアプローチできる実験系は限られている。この為、筆者らの光遺伝学ALSモデルは、期せずして、TDP-43凝集の上流のイベントを研究するための貴重なモデルとなるのではないか、と考えている。
光遺伝学ALSモデルの応用:TDP-43とDNA損傷応答
ALSで蓄積する封入体の主成分としてTDP-43が同定されて以来、TDP-43凝集体の物性に大きな注目が集まってきた。しかし、筆者らの研究も含めて、凝集体形成に付随しないTDP-43の細胞毒性が、独立に報告され始めており[6]、今後は、正常細胞において細胞核に豊富に局在するTDP-43が、如何にして細胞質に移行するのか?という凝集前の過程を理解することが、より初期のALS病態を理解するために重要性を増してくると予想される。筆者らは、CRY2oligを用いてTDP-43の分子間相互作用を促進することで、TDP-43の細胞質への移行が促進されることを見出した。この興味深い現象のメカニズムは、現在、全く未知である。ALSにおいて、あたかも“青色光”と同様の効果を発揮し、TDP-43の病理的な相転移や細胞質への移行を促進する生体内の因子が存在するとすれば、それはなんなのだろうか?
この問題に挑戦するためには、必然的に正常細胞の細胞核におけるTDP-43の機能を深く理解しなくてはならない。筆者らは、その一つとして、DNA損傷応答におけるTDP-43の役割に着目している。興味深いことに、TDP-43の枯渇は、DNA二重鎖切断の蓄積を招き、対照的に、TDP-43の過剰発現は、DNA損傷に対する保護効果が認められる[7-10]。これらの遺伝学的研究は、TDP-43がDNAの損傷応答に関与していることを示唆している。筆者らは、これらのクラシカルな遺伝学的な知見を一歩前進させ、TDP-43の相転移がDNA損傷の原因なのか、あるいは、様々なゲノムストレスによって誘発されるDNA損傷を修復する役割を担っているのか、という点を、opTDP-43の光操作を用いて明らかにしていきたいと考えている。ゼブラフィッシュは、神経活動に伴って起こるDNA損傷が及ぼす影響を生体レベルで研究する系として力を発揮し始めており[11, 12]、ゼブラフィッシュ光遺伝学ALSモデルが、いまだ未知である、DNA損傷とALSの関係性の解明に貢献することが期待される。
謝辞
本研究を実施するにあたりご助言をくださった、京都大学iPS細胞研究所、今村恵子博士、井上治久博士、東京医科大学、郭伸先生に深く感謝いたします。本研究は、せりか基金、「生命の彩」ALS研究助成金、加藤記念難病研究助成基金, 第一三共生命科学研究振興財団、武田科学振興財団、科研費、NBRP/AMEDの支援を受けて行われました。
引用文献
1. Asakawa, K., H. Handa, and K. Kawakami, Optogenetic modulation of TDP-43 oligomerization accelerates ALS-related pathologies in the spinal motor neurons. Nat Commun, 2020. 11(1): p. 1004.
2. Arai, T., et al., TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun, 2006. 351(3): p. 602-11.
3. Neumann, M., et al., Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science, 2006. 314(5796): p. 130-3.
4. Asakawa, K., et al., Genetic dissection of neural circuits by Tol2 transposon-mediated Gal4 gene and enhancer trapping in zebrafish. Proc Natl Acad Sci U S A, 2008. 105(4): p. 1255-60.
5. Taslimi, A., et al., An optimized optogenetic clustering tool for probing protein interaction and function. Nat Commun, 2014. 5: p. 4925.
6. Arnold, E.S., et al., ALS-linked TDP-43 mutations produce aberrant RNA splicing and adult-onset motor neuron disease without aggregation or loss of nuclear TDP-43. Proc Natl Acad Sci U S A, 2013. 110(8): p. E736-45.
7. Hill, S.J., et al., Two familial ALS proteins function in prevention/repair of transcription-associated DNA damage. Proc Natl Acad Sci U S A, 2016. 113(48): p. E7701-E7709.
8. Mitra, J., et al., Motor neuron disease-associated loss of nuclear TDP-43 is linked to DNA double-strand break repair defects. Proc Natl Acad Sci U S A, 2019. 116(10): p. 4696-4705.
9. Konopka, A., et al., Impaired NHEJ repair in amyotrophic lateral sclerosis is associated with TDP-43 mutations. Mol Neurodegener, 2020. 15(1): p. 51.
10. Giannini, M., et al., TDP-43 mutations link Amyotrophic Lateral Sclerosis with R-loop homeostasis and R loop-mediated DNA damage. PLoS Genet, 2020. 16(12): p. e1009260.
11. Zada, D., et al., Sleep increases chromosome dynamics to enable reduction of accumulating DNA damage in single neurons. Nat Commun, 2019. 10(1): p. 895.
12. Zada, D., et al., Parp1 promotes sleep, which enhances DNA repair in neurons. Mol Cell, 2021. 81(24): p. 4979-4993 e7.
著者
浅川和秀1、2、半田宏1、川上浩一2
1、東京医科大学 ケミカルバイオロジー講座、 2、国立遺伝学研究所 発生遺伝学研究室
